About us
Research areas
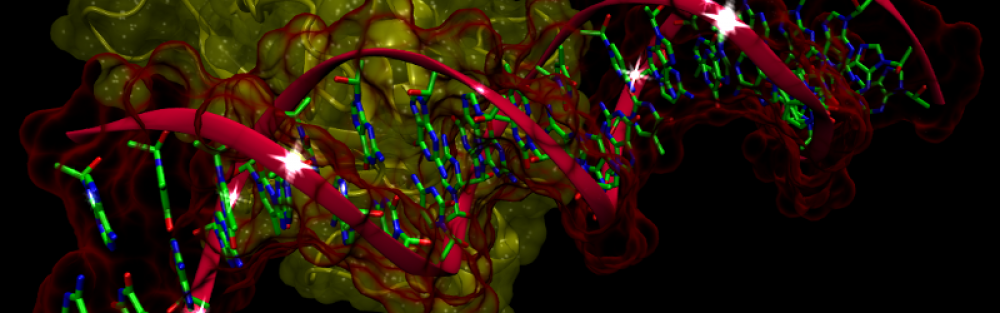
Pharmacogenomics: This integrated approach has the potential to dramatically reduce drug adverse reactions and increase therapeutic effects by correlating knowledge annotated genes, proteins, and single nucleotide polymorphisms with drug behavior. Establishing the connections between drugs, molecular pathways and diseases is essential to accelerate drug discovery and to facilitate the development of safer therapeutics. Primary applications include the detection of the most perturbed disease-related pathways and sub-networks, identification of direct molecular targets, prediction of drug mode of action, characterization of drug-drug interactions in multi-drug therapy, and rational drug repositioning. Finally, the correlation of genotypes and drug response phenotypes is thought to be the key to creating personalized drugs, viz. tailor-made pharmaceuticals adapted to each person's own genetic makeup.
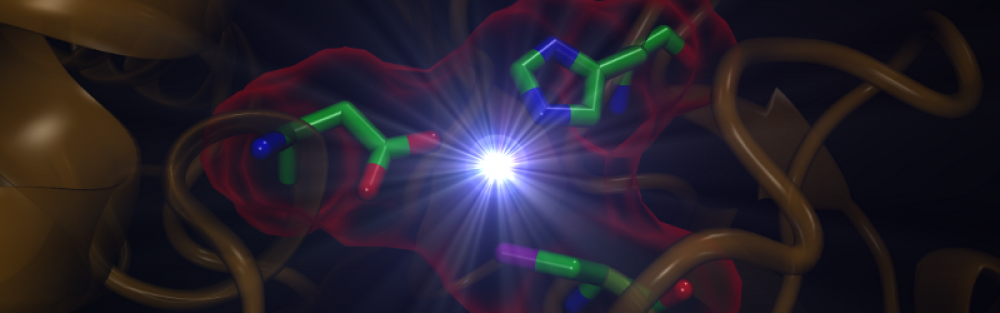
Network Pharmacology: Protein-drug interactome is of paramount importance for drug discovery, a field that is facing significant difficulties related to the binding promiscuity of drugs and the intricate networks of drug-target interactions, far more complicated than originally anticipated. Polypharmacology, also referred to as Network Pharmacology, rapidly emerges as a new trend in pharmaceutical research to challenge the problem of drug resistance and side-effects caused by compounds with unanticipated promiscuous binding properties. By tuning up molecular selectivity profiles, polypharmacology concentrates on multi-target drugs, which are selectively non-selective, i.e. target disease-related pathways or sub-networks rather than individual proteins.

Network Biology: In a crowded cellular environment, numerous interactions occur between molecular species in a cell. They interact with each other in specific ways to perform their biological functions. Although the classical view of molecular interactions arranges them into biological pathways, which are often treated as independent functional entities, recent functional genomic experiments reveal very extensive cross-talks between pathways, suggesting a much more convoluted picture of molecular interactions in vivo. These inter-pathway connections are currently the subject of intense research since they are responsible for the extreme complexity of biological systems, yet are still poorly defined at the proteome level. It is hoped that the emergence of systems-level disciplines, such as Network Biology, will help uncover the organizing principles of the intercellular webs of interactions, which collectively control the behavior of a cell.

Cheminformatics: Qualitative protein function annotation is typically followed by a comprehensive functional characterization at the molecular level. In particular, combining the evolutionary information with structure-based docking holds a significant promise to improve drug discovery and design. The intensive research in this field culminated in the development of eSimDock, which employs structural information extracted from weakly related proteins to perform rapid similarity-based ligand docking and binding affinity prediction, and GeauxDock, a novel mixed-resolution ligand docking and ranking approach. Both tools fit perfectly the scope of biological network analysis, since they are generally applicable to large-scale projects focused on the identification of potential small molecule-protein interactions across the entire proteomes.
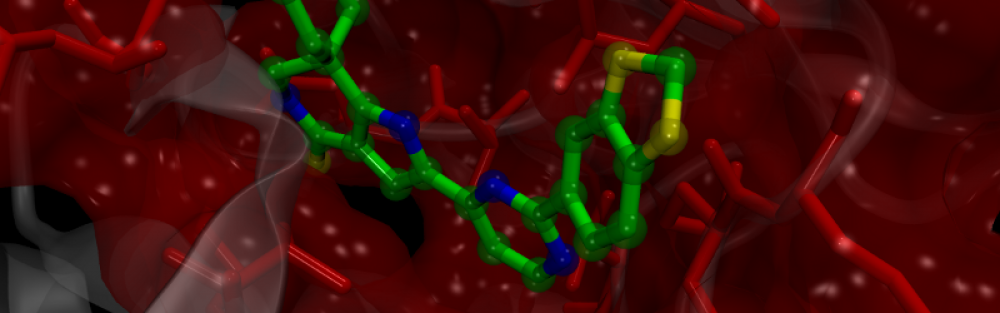
Functional Genomics: Powerful structure-based approaches to protein function inference effectively utilize low-to-moderate quality protein models for function assignment and are of considerable practical assistance in the construction and analysis of biological pathways and complex molecular networks. A number of innovative computational approaches to indexing ligand-protein interactions have been developed to date, including eFindSite, a highly accurate algorithm for ligand binding site prediction and ligand-based virtual screening. Undoubtedly, precise functional annotation is essential for further studies on interactions between proteins and other molecular species in a cell. In our research on biological networks, we apply a diverse collection of highly accurate tools for function inference to cover most of the functional aspect of proteins, including protein-nucleic acid, protein-ligand, protein-metal and protein-protein interactions.

Structural Bioinformatics: Systems Biology seeks to understand how the components of complex living systems interact and how their malfunction causes disease. Genome sequencing has provided the community with the vast amount of sequence information, however, the functions of the majority of gene products remain unknown. To extend functional inference approaches to low levels of sequence identity, structure-based methods are widely used. Consequently, protein structure prediction, particularly template-based approaches, are the first step in across-proteome function inference. We approach the problem of biological network analysis from a very structure-oriented perspective. We attempt to quantify the interactions between cellular entities at the molecular level by applying a collection of the state-of-the-art modeling techniques, such as eThread, to construct the molecular structures of all gene products. Such strategy maximizes the coverage of a given proteome by functionally relevant conformations, which is critical for proteome-wide function annotation.
Research projects
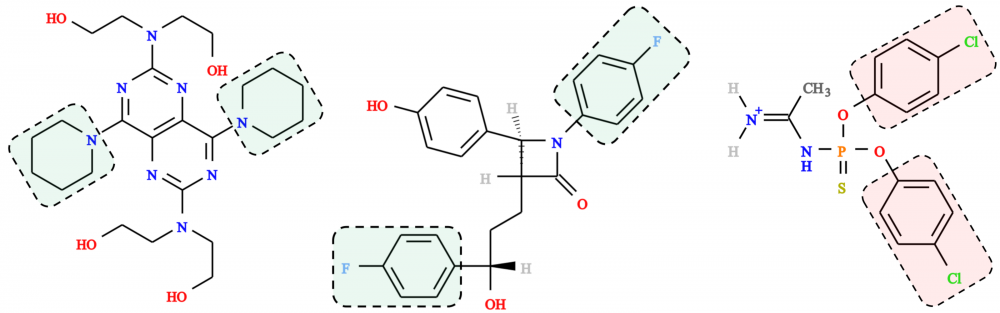
Prediction of toxicity and synthesizability: One of the increasingly important component of modern drug discovery is the prediction of the toxicity of drug candidates. In collaboration with Supratik Mukhopadhyay and Hsiao-Chun Wu, we developed eToxPred, a new approach to reliably estimate the toxicity and synthetic accessibility of small organic compounds. eToxPred employs machine learning algorithms trained on molecular fingerprints to evaluate drug candidates. Encouragingly, it predicts the synthetic accessibility with the mean square error of only 4% and the toxicity with the accuracy of as high as 72%. eToxPred is a valuable tool that can be employed at the outset of drug discovery to filter out those drug candidates that are potentially toxic or would be difficult to synthesize. read more
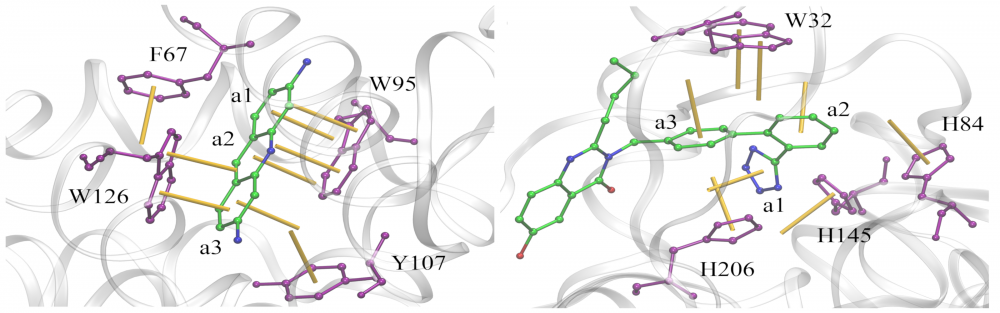
Analysis of aromatic interactions: Aromatic stacking has long been recognized as one of the key constituents of ligand-protein interfaces. We developed a two-parameter geometric model to analyze aromatic contacts in the experimental and computer-generated structures of ligand-protein complexes, considering various combinations of aromatic amino acid residues and ligand rings. Although modeling aromatic stacking with van der Waals and Coulombic potentials generally provides a sufficient specificity, the geometry of π-π contacts in high-scoring docking conformations could still be improved. The comprehensive analysis of aromatic geometries at ligand-protein interfaces lies the foundation for the development of type-specific statistical potentials to more accurately describe aromatic interactions in molecular docking. read more
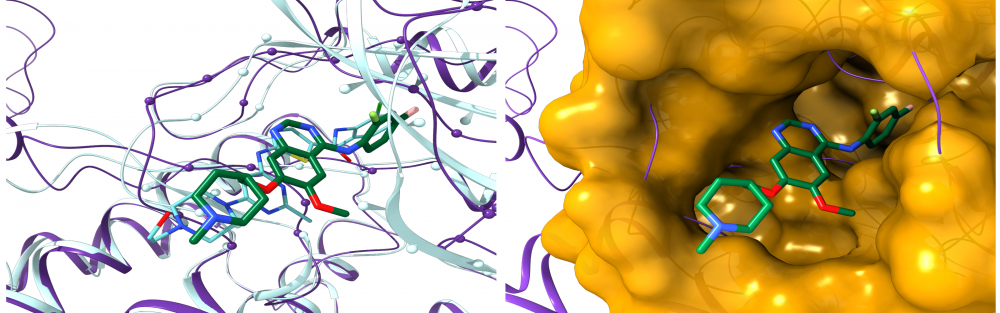
Drug repositioning for orphan diseases: About 7,000 rare, or orphan, diseases affect more than 350 million people worldwide. Although these conditions collectively pose significant health care problems, drug companies seldom develop drugs for orphan diseases due to extremely limited individual markets. Computer-aided drug repositioning is a cheaper and faster alternative to traditional drug discovery offering a promising venue for orphan drug research. We developed eRepo-ORP, a comprehensive resource constructed by a large-scale repositioning of existing drugs to orphan diseases with eThread, eFindSite and eMatchSite. A systematic exploration of 320,856 possible links between known drugs in DrugBank and orphan proteins obtained from Orphanet revealed as many as 18,145 candidates for repurposing. read more
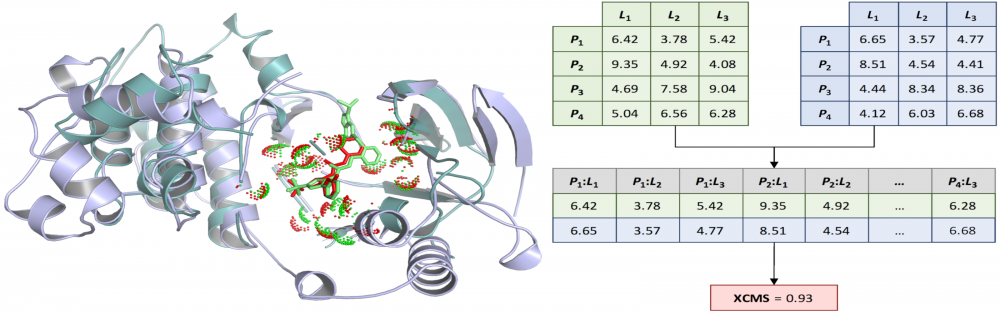
Similarity measures for ligand-binding conformations: Structural and computational biologists often need to measure the similarity of ligand binding conformations. The commonly used root-mean-square deviation (RMSD) is not only ligand-size dependent, but also may fail to capture biologically meaningful binding features. To address these issues, we developed the Contact Mode Score (CMS), a new metric to assess the conformational similarity based on intermolecular protein-ligand contacts. The CMS is less dependent on the ligand size and has the ability to include flexible receptors. In order to effectively compare binding poses of non-identical ligands bound to different proteins, we further developed the eXtended Contact Mode Score (XCMS). CMS and XCMS provide a meaningful assessment of the similarity of ligand binding conformations. read more
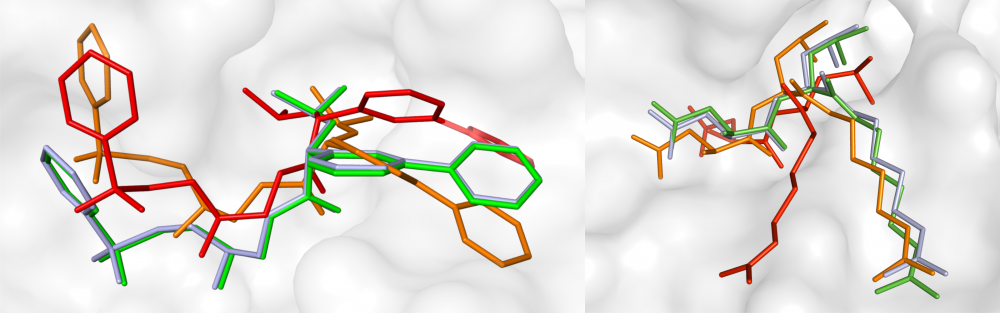
Mixed-resolution drug docking: Molecular docking is an important component of computer-aided drug discovery. As part of a collaborative effort within LA-SiGMA, we developed GeauxDock, a new docking approach that builds upon the ideas of ligand homology modeling. GeauxDock features a descriptor-based scoring function integrating evolutionary constraints with physics-based energy terms, a mixed-resolution molecular representation of protein-ligand complexes, and an efficient Monte Carlo sampling protocol. In order to drive docking simulations towards experimental conformations, the scoring function was carefully optimized to produce a correlation between the total pseudo-energy and the native-likeness of binding poses. read more
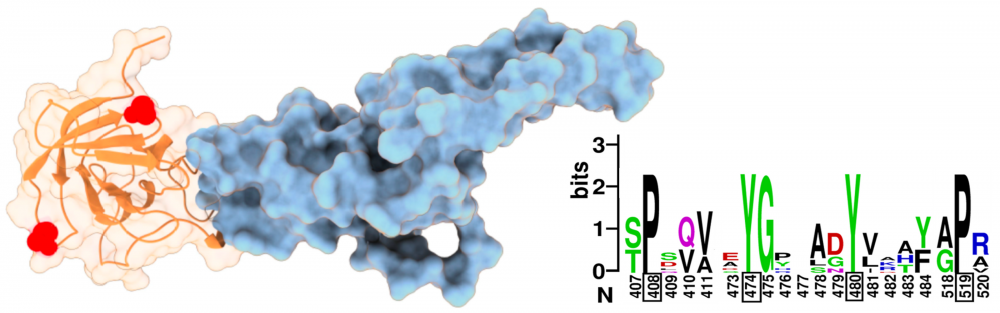
Structure and function of gK: Alphaherpesviruses are a subfamily of herpesviruses that include the significant human pathogens herpes simplex viruses (HSV). HSV-1 glycoprotein K (gK) is a multi-membrane spanning virion glycoprotein essential for virus entry into neuronal axons, virion assembly, and pathogenesis. However, little is known about which gK domains and residues are most important for maintaining these functions across all alphaherpesviruses. We collaborate with Gus Kousoulas to elucidate the important structural features of gK that are involved in gK-mediated regulation of virus-induced membrane fusion. A greater understanding of mechanisms governing alphaherpesvirus membrane fusion is expected to inform the rational design of therapeutic and prevention strategies to combat herpesviral infection and pathogenesis. read more
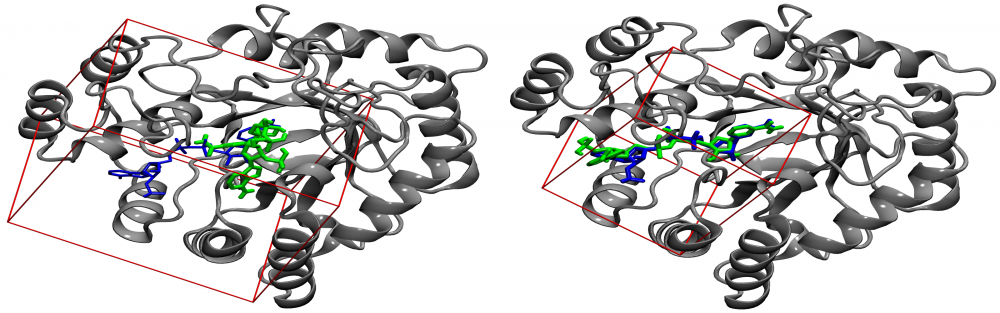
Optimization of docking parameters: Molecular docking has profound applications in drug discovery and development. One of the critical parameters for ligand docking is the size of a search space used to identify low-energy binding poses of drug candidates. We proposed a new procedure for calculating the optimal docking box size that maximizes the accuracy of binding pose prediction and yields an improved ranking in virtual screening. Importantly, the optimized search space systematically gives better results than the default method not only for experimental pockets, but also for those predicted from protein structures. Our approach can be employed to fully automate large-scale virtual screening calculations by customizing docking protocols on the fly for individual library compounds. read more
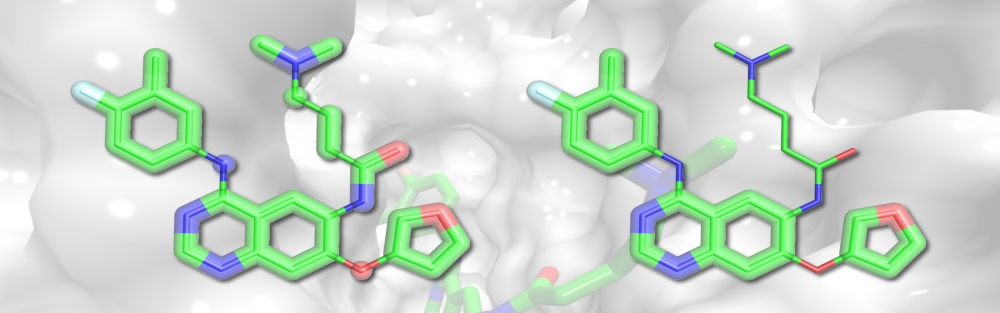
Molecular fragmentation and synthesis: The trend in library design for virtual screening has shifted to produce screening collections specifically tailored to modulate the function of a particular target or a protein family. In collaboration with Supratik Mukhopadhyay, we developed eMolFrag, a new software to decompose organic compounds into non-redundant fragments retaining molecular connectivity information. These building blocks can subsequently be employed by eSynth, an exhaustive graph-based search algorithm to computationally synthesize new compounds. Based on a small set of already developed bioactives, this approach is capable of generating diverse collections of molecules with the desired activity profiles. read more
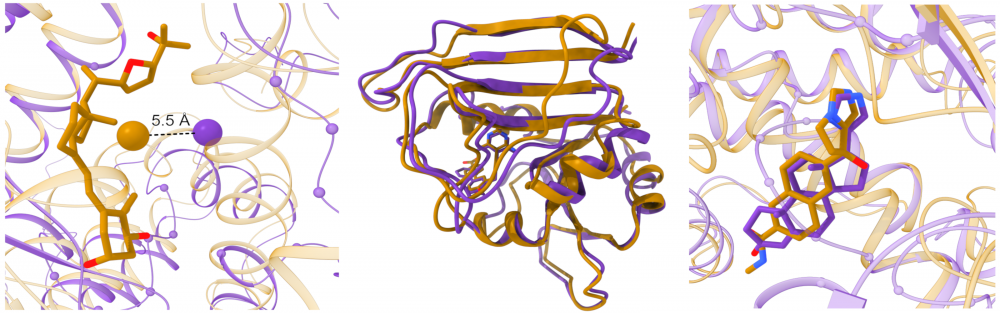
Large-scale modeling of protein-drug interactions: The structural information on proteins in their ligand-bound conformational state is invaluable for protein function studies and rational drug design. However, the repertoire of the experimentally determined structures of holo-proteins is not only limited, but also these structures do not always include pharmacologically relevant compounds at their binding sites. To complement the existing repositories, we created eModel-BDB, a database of 200,005 comparative models of drug-bound proteins based on interaction data obtained from the Binding Database. Furthermore, we collaborate with Lukasz Kurgan to provide the Protein-Drug Interaction Database comprising a large number of putative protein-drug interactions that cover the entire structural human proteome. read more
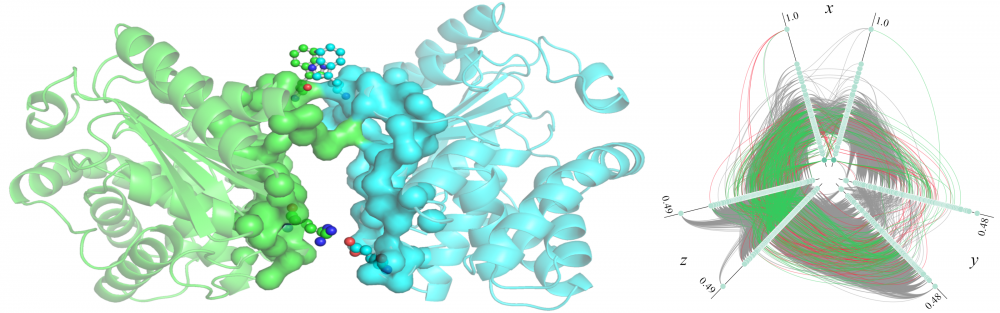
Modeling of protein-protein interactions: The identification of protein-protein interactions (PPIs) is vital for understanding protein function, elucidating interaction mechanisms, and for practical applications in drug discovery. To improve the state-of-the-art in PPI modeling, we developed eFindSitePPI to predict binding residues in a target protein structure with machine learning, and eRankPPI to identify near-native conformations generated by protein docking with a new scoring function utilizing interface probability estimates and a contact-based symmetry score. Furthermore, we devised a high-throughput protocol for the bottom-up assembly of protein interaction networks based on all-to-all protein docking. These tools can be used to reliably identify and model biologically relevant protein assemblies at the proteome scale. read more
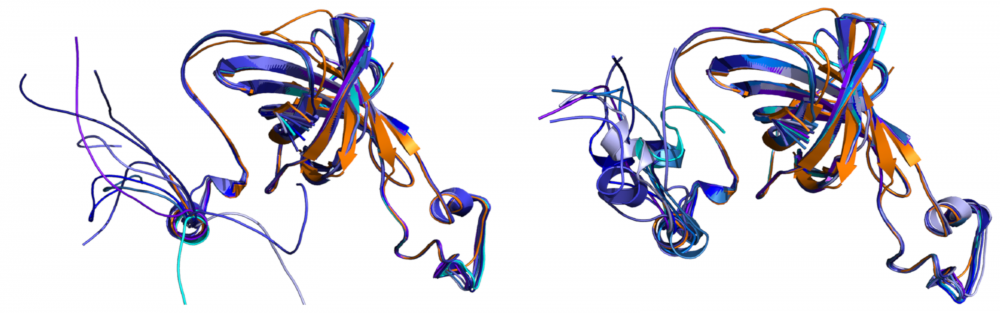
Structure and function of photosystem II: In higher plant Photosystem II, the PsbO, PsbP and PsbQ proteins provide critical support for oxygen evolution at physiological calcium and chloride concentrations. We collaborate with Terry Bricker to examine the structure of these components when bound to Photosystem II. This interdisciplinary project combines protein crosslinking, radiolytic footprinting coupled with high resolution tandem mass spectrometry, and molecular modeling to provide structural and functional information regarding the organization of the higher plant photosystem. read more
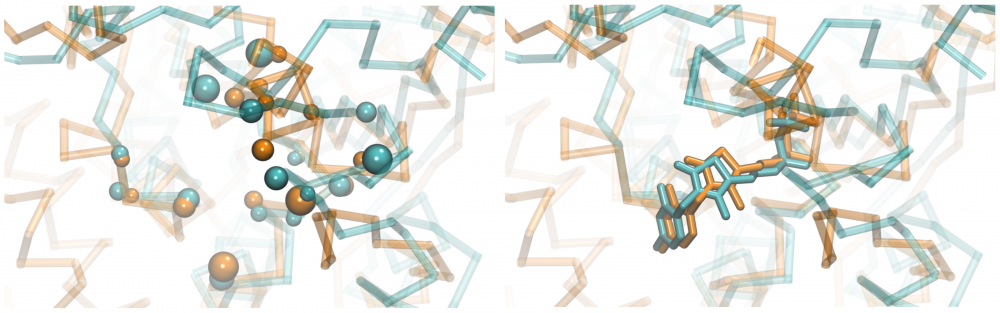
Ligand-binding site alignment: Detecting similarities between ligand binding sites in the absence of global homology between target proteins has been recognized as one of the critical components of modern drug discovery. Towards this goal, we developed eMatchSite, a new method to construct sequence order-independent alignments of ligand-binding sites with the Hungarian algorithm and machine learning. eMatchSite not only outperforms other approaches to match binding sites, but it also offers a remarkably high tolerance to structure distortions in protein models. Constructing biologically correct alignments opens up the possibility to investigate drug-protein interaction networks for complete proteomes with prospective systems-level applications in polypharmacology and rational drug repositioning. read more
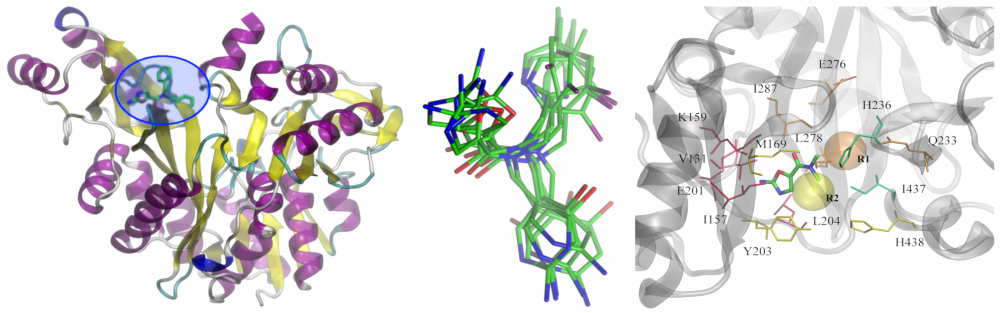
Redesign of biotin carboxylase inhibitors: As the frequency of antibiotic resistant bacteria steadily increases, there is an urgent need for new antibacterial agents. Because fatty acid synthesis is only used for membrane biogenesis in bacteria, the enzymes in this pathway are attractive targets for antibacterial development. Amino-oxazole inhibits biotin carboxylase (BC) activity in Gram-negative organisms. In collaboration with Grover Waldrop, we redesigned previously identified lead inhibitors of BC to expand the spectrum of bacteria sensitive to the amino-oxazole derivatives by including Gram-positive species. Structural insights into drug-BC interactions will be exploited to increase the potency of amino-oxazole inhibitors towards both Gram-negative as well as Gram-positive species. read more
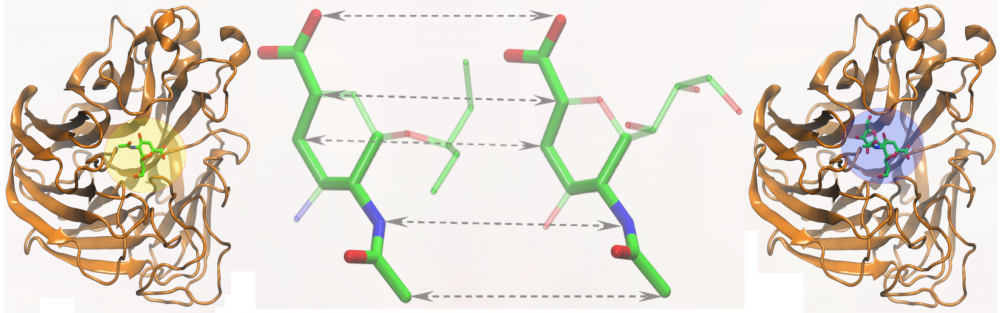
Similarity-based ligand docking: A common strategy for virtual screening considers a systematic docking of a large library of organic compounds into the target sites in protein receptors. We developed eSimDock, a new approach to ligand docking and binding affinity prediction. This algorithm employs non-linear machine learning-based scoring functions to improve the accuracy of ligand ranking, and similarity-based binding pose prediction to increase the tolerance to structural imperfections in the target structures. Comprehensive benchmarking calculations demonstrated that the performance of eSimDock is largely unaffected by the deformations of ligand binding regions, thus it represents a practical strategy for across-proteome virtual screening using protein models. read more
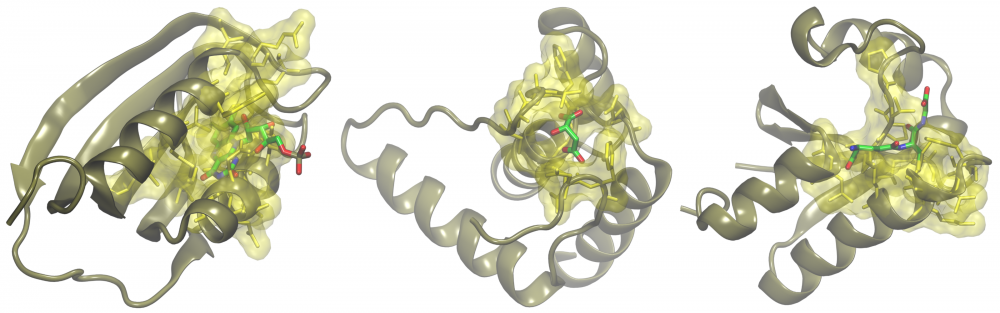
Annotation of small proteins: A growing body of evidence shows that gene products encoded by short open reading frames play key roles in numerous cellular processes. Yet, they are generally overlooked in genome assembly, escaping annotation because small protein-coding genes are difficult to predict computationally. There are still a considerable number of small proteins whose functions are yet to be characterized. To address this issue, we applied a collection of structural bioinformatics algorithms to infer molecular function of putative small proteins from the mouse proteome. Our results strongly indicate that many small proteins adopt three-dimensional structures and are fully functional, playing important roles in transcriptional regulation, cell signaling and metabolism. read more
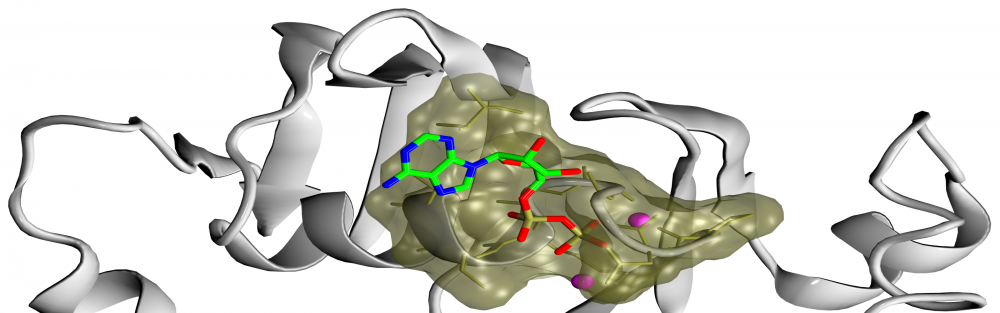
Ligand-binding site prediction: Much needed functional annotation of gene products across different species often requires the knowledge of protein-ligand interactions. Towards this goal, we developed eFindSite, an algorithm to efficiently identify ligand-binding sites and residues from weakly homologous templates with highly sensitive meta-threading, improved clustering techniques, and advanced machine learning methods. eFindSite can also be used to conduct ligand-based virtual screening employing consensus molecular fingerprints. Carefully calibrated confidence estimates strongly indicate that highly reliable ligand binding predictions are made for the majority of gene products in a given proteome, thus eFindSite holds a significant promise for large-scale genome annotation and drug development projects. read more
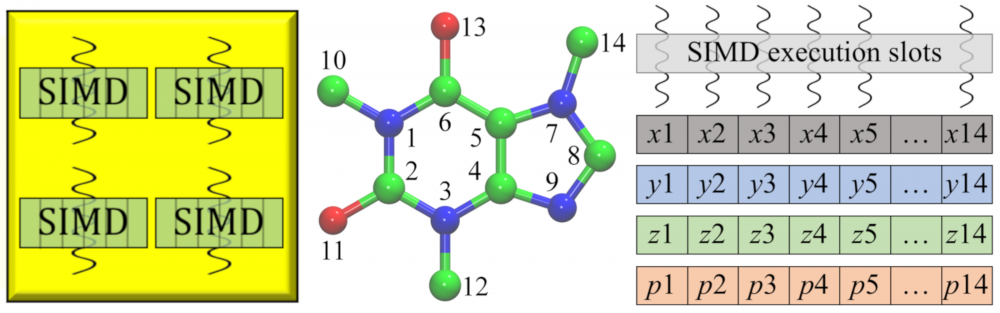
High-performance computing: Drug development is routinely streamlined using computational approaches to improve hit identification and lead selection, enhance bioavailability, and reduce toxicity. New challenges arose because processing a large volume of data demands unprecedented computing resources. As part of a collaborative effort within LA-SiGMA, we ported several of our codes to heterogeneous computing platforms. For instance, GeauxDock can be deployed on multi-core Central Processing Units (CPUs) as well as massively parallel accelerators, Intel Xeon Phi and NVIDIA Graphics Processing Unit (GPU). Further, a parallel version of eFindSite was implemented mainly for the Intel Xeon Phi platform. These parallel codes yield significant performance improvements considerably accelerating their large-scale applications. read more
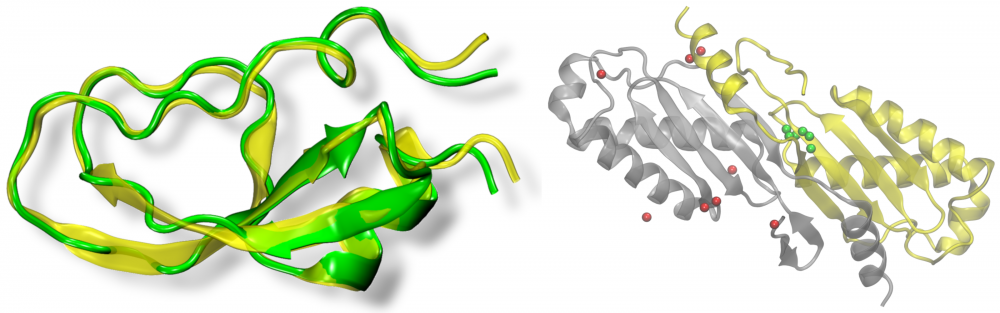
Protein sequence optimization: Many structural bioinformatics approaches employ sequence profile-based threading algorithms. To improve fold recognition rates, homology searching may include artificially evolved amino acid sequences. We developed eVolver, an optimization engine that evolves protein sequences to stabilize the respective structures by a variety of potentials. Sequences generated by eVolver have high capabilities to recognize the correct protein structures using standard sequence profile-based techniques, thus they can be incorporated into existing threading approaches to increase their sensitivity. These sequences also provide a linkage between protein structure and function in computer simulations focused on the study of the completeness of protein structure space. read more
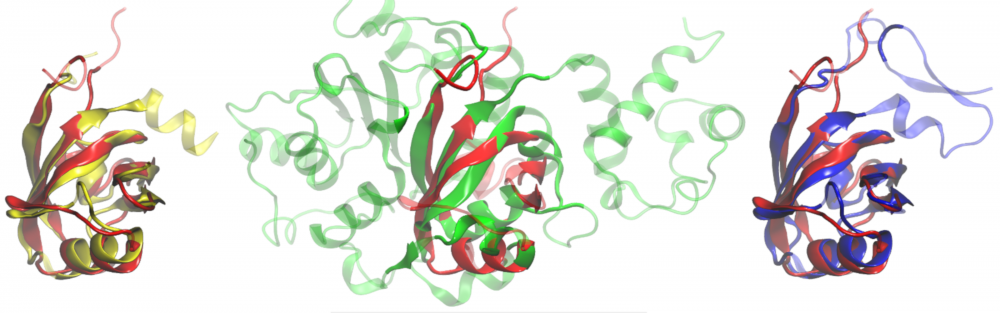
Protein structure modeling: Template-based modeling that employs various meta-threading techniques is currently the most accurate, and consequently the most commonly used, approach to predict protein structures. We developed eThread, a highly accurate meta-threading procedure to identify structural templates followed by the construction of the corresponding target-template alignments and 3D models. We also extended the functionality of eThread to select functional templates covering a broad spectrum of protein molecular function, including ligand, metal, inorganic cluster, protein and nucleic acid binding. We demonstrated that in addition to accurate protein structure modeling, meta-threading effectively detects many facets of molecular function, even in a low sequence identity regime. read more
Quick intro
Computational Systems Biology aims to develop and apply efficient algorithms to address critical scientific questions through computer simulations and theoretical modeling. The system-wide modeling is particularly relevant in modern biological sciences, where the key challenge has shifted from the study of single molecules to the exhaustive exploration of molecular interactions and biological processes at the level of complete proteomes.