Ligand-binding site alignment
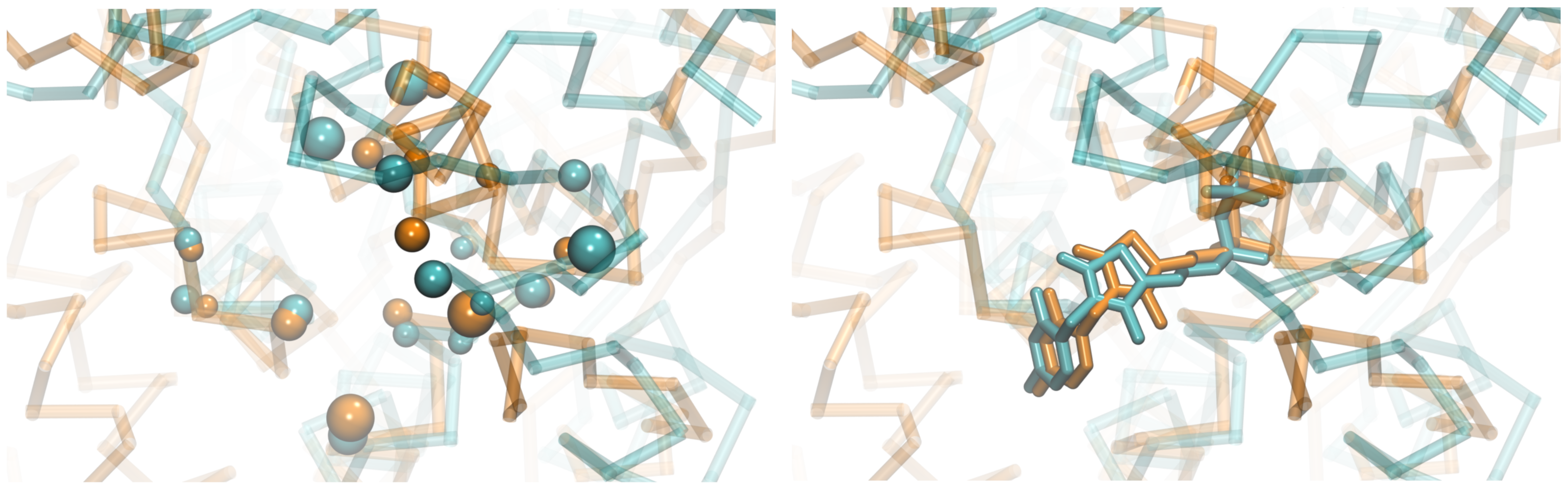
Detecting similarities between ligand binding sites in the absence of global homology between target proteins has been recognized as one of the critical components of modern drug discovery. Towards this goal, we developed eMatchSite, a new method to construct sequence order-independent alignments of ligand-binding sites with the Hungarian algorithm and machine learning. eMatchSite not only outperforms other approaches to match binding sites, but it also offers a remarkably high tolerance to structure distortions in protein models. Constructing biologically correct alignments opens up the possibility to investigate drug-protein interaction networks for complete proteomes with prospective systems-level applications in polypharmacology and rational drug repositioning.