Analysis of aromatic interactions
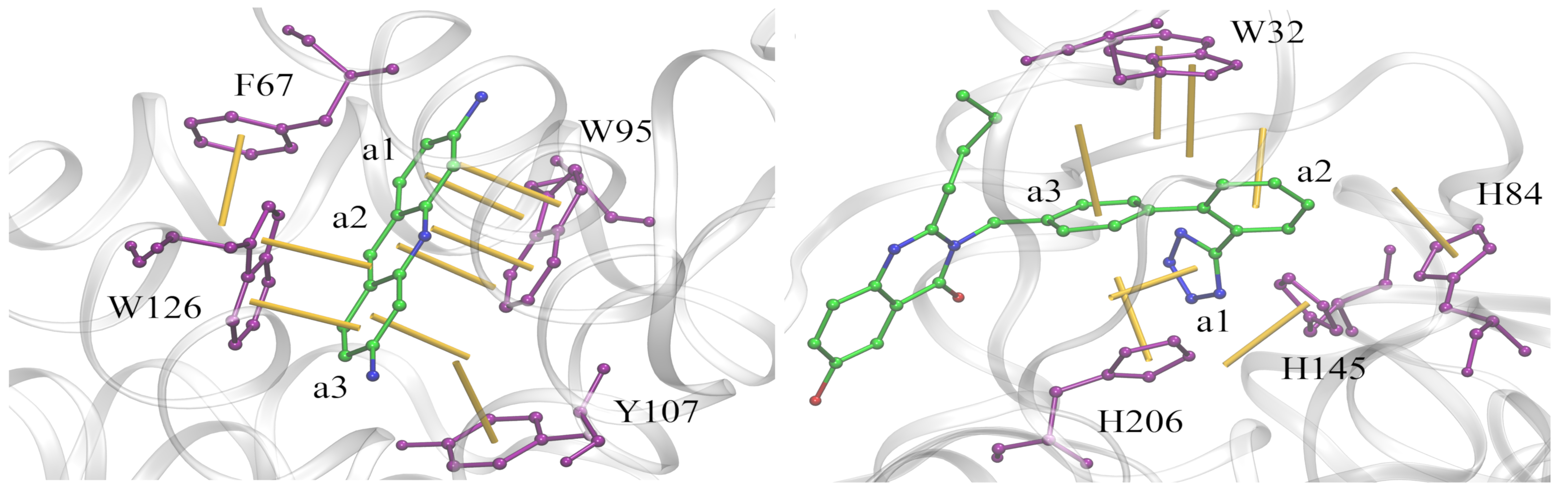
Aromatic stacking has long been recognized as one of the key constituents of ligand-protein interfaces. We developed a two-parameter geometric model to analyze aromatic contacts in the experimental and computer-generated structures of ligand-protein complexes, considering various combinations of aromatic amino acid residues and ligand rings. Although modeling aromatic stacking with van der Waals and Coulombic potentials generally provides a sufficient specificity, the geometry of π-π contacts in high-scoring docking conformations could still be improved. The comprehensive analysis of aromatic geometries at ligand-protein interfaces lies the foundation for the development of type-specific statistical potentials to more accurately describe aromatic interactions in molecular docking.